分光法による分子構造決定において、古くからIRスペクトルが重要な役割を果たしてきたことは、IRの章で書いたとおりですが、当然、IRだけでは完全に有機分子の構造を決定することは困難であることが多いのは言うまでもありません。IRが化学結合を見ているのに対し、より新しい分光法であるNMR(Nuclear Magnetic Resonance:核磁気共鳴)は「原子核の磁気的・電子的環境」を観測するものです。構成原子がどういう状態にあるかを、直接見ることができるNMRは、IRに比べて遥かに具体的な情報が得られ、複雑な分子の構造決定に大いに役立ちます。タンパク質の構造決定にさえ、NMRは利用されています。これはIRではまず不可能です。
NMRスペクトルは、分子を強い磁場の中に置き、マイクロ波の吸収を見たものです。原子核の周りの電子の状態(密度や広がりなど)によって磁場に対する反応が異なり、それが即ち吸収パターンの違いとなって現れます。1H及び13C-NMRはテトラメチルシラン(TMS)を基準とし、TMSとの吸収位置のずれで比較を行います。最新のGAMESSでは、NMRの絶対遮蔽定数を計算することができ、TMSの絶対遮蔽定数との差から化学シフトを計算で求めることができます。果たして、どのくらいの精度で計算することができるのでしょうか。
化学シフト計算

MOPACでは化学シフトの計算を行うことはできません。GAMESSでも比較的最近実装されたもので、PC-GAMESSでは計算できません。WinGAMESSの最新版で計算を行うことができます。WinmostarとWinGAMESSを使い、アセトアルデヒドの1H及び13C-NMRの化学シフトを求めてみましょう。
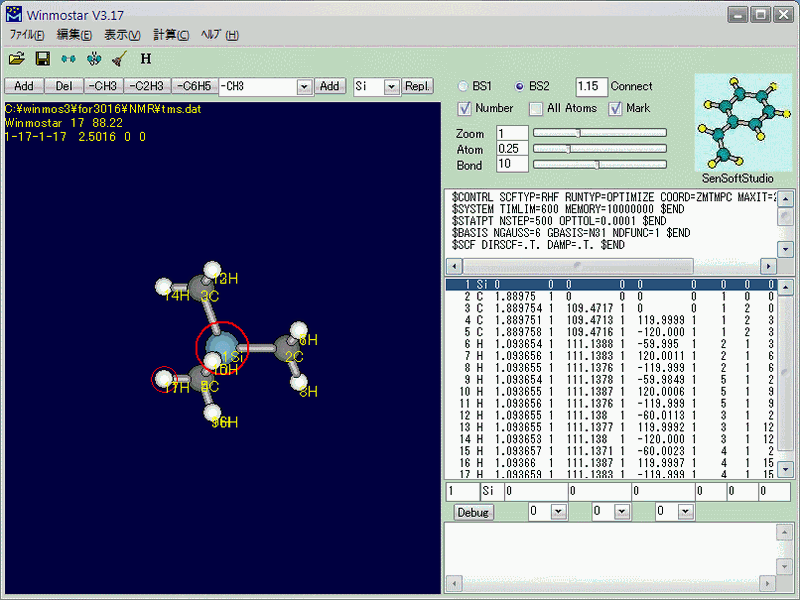
まず最初に、TMSの絶対遮蔽定数を求めなくてはなりません。TMSをモデリングしたら(このモデリングは簡単なので省略)、キーワードとして「PM3 EF PRECISE」を入力し(Fig.1)、MOPACで最適化を行います。 最終的にはGAMESSで最適化を行いますが、より良い初期構造を与えることで総合的な計算時間の短縮を図るため、先にMOPACで最適化をします。
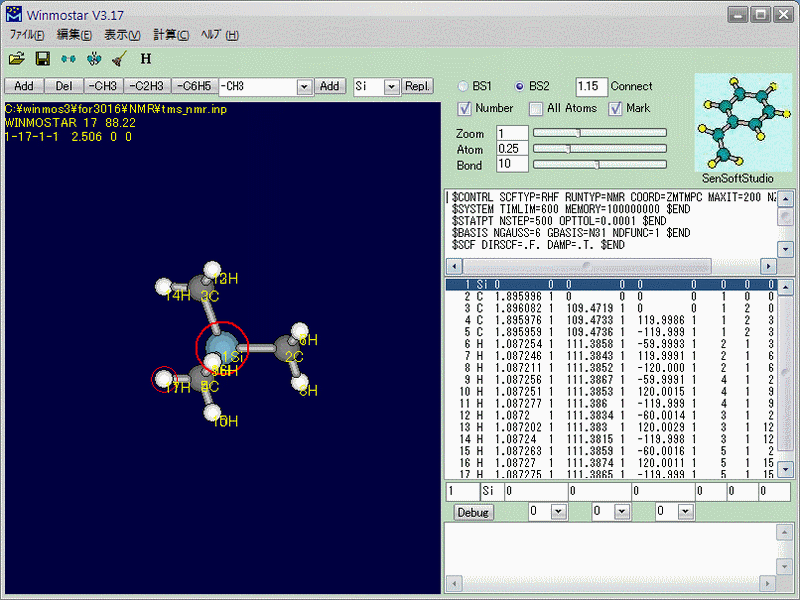
最適化が終わったら、今度はGAMESSの入力を作成します。[計算(C)]→[GAMESSパラメータをセット]でキーワードを仮入力し、$BASISセクションを「NGAUSS=6 GBASIS=N31 NDFUNC=1」(つまりRHF/6-31G(d))に書き換えます(Fig.2)。計算を実行し、終了したら[計算(C)]→[Import out]から最適化構造を読み込みます。
ここからが、本題のNMR遮蔽定数の計算です。キーワード欄の、$CONTRLセクションのRUNTYPをNMRとし、$SCFセクションのDIRSCFを.F.に書き換えます(Direct SCFでは計算できません)(Fig.3)。
GAMESSのNMR計算はGIAO法で行われます。適当な名前で保存し、計算を実行します。この計算は非常に時間がかかります(Athlon64 3000+で3時間)。計算が終了したら、出力ファイル(.out)をテキストエディタで開きます([計算(C)]→[Edit out(log)])。
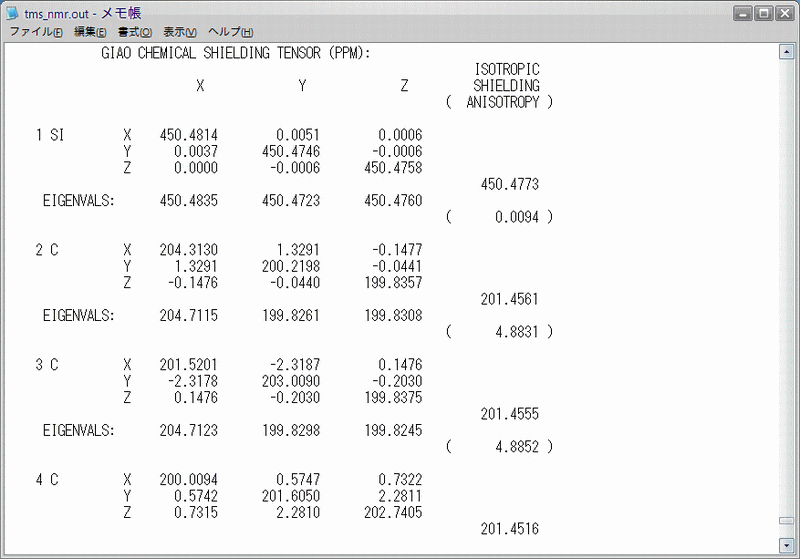
NMRの計算の出力は、.outファイルの一番最後にあります(Fig.4)。一番右の「ISOTROPIC SHIELDING(等方性遮蔽)」の値が見るべき値です。水素は12個、炭素は4個ありますが、実際のNMRで観測されるのはそれらの平均値ですので、出力された値の平均値を計算値とします。ちなみに私が行った計算では、水素原子の絶対遮蔽定数が32.8961 ppm,炭素原子が201.4536 ppmでした。この値は、構造最適化:RHF/6-31g(d)/NMR:RHF/6-31g(d)での値ということになります。これと同じモデル化学で他の有機分子の遮蔽定数を求め、TMSとの差をとることにより、化学シフトの計算を行うことができます。
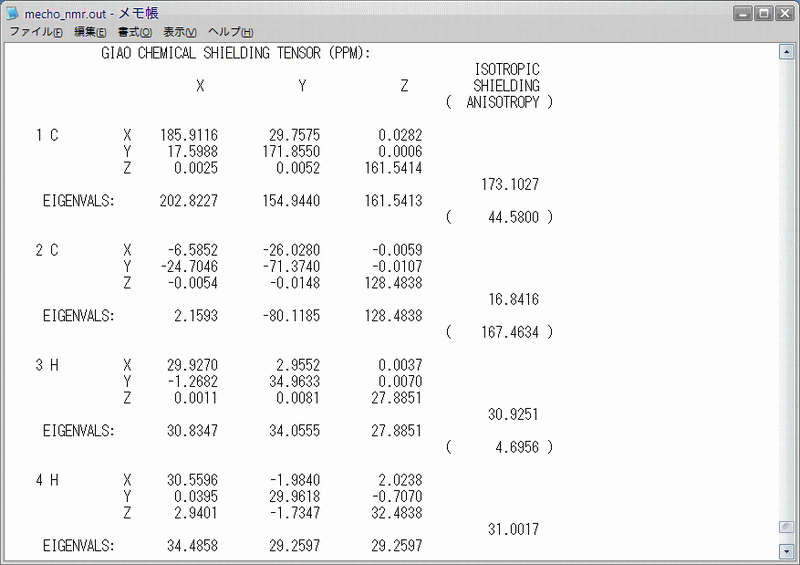
アセトアルデヒドについて、TMSと同じ手順で計算を行うと、Fig.5のような計算結果が得られます。私が行った計算による絶対遮蔽定数は、アルデヒド水素が23.3236 ppm,メチル水素が30.9764 ppm,カルボニル炭素が16.8416 ppm,メチル炭素が173.1027 ppmです。それぞれの値を先ほど求めたTMSの絶対遮蔽定数から引くと、化学シフトはそれぞれ9.5725 ppm,1.9197 ppm,184.6120 ppm,28.3509 ppmとなります。ちなみに、実測値はそれぞれ9.789 ppm,2.206 ppm,199.93 ppm,30.89 ppmです。どうでしょう。なかなか良い一致を示しているように思います。
計算精度
さて、一例としてアセトアルデヒドの化学シフトを求めてみましたが、これだけでは本当に精度が高い計算なのかどうかはわかりません。そこで、いろいろな分子について計算し、重クロロホルム中の実測値との比較を行ってみました。尚、実測値は産総研有機化合物スペクトルデータベース[SDBSWeb: http://www.aist.go.jp/RIODB/SDBS/ (National Institute of Advanced Industrial Science and Technology, 2005/07/17-24)]より引用しました。また、シクロプロパンの13C化学シフトはhttp://chembionews.cambridgesoft.com/art_print.cfm?S=233より引用しました。
| Compounds | Nucleus | Obs.(ppm) | Calc.(ppm) | diff. |
|---|---|---|---|---|
| methanol |
H(a) | 3.417 | 3.345 | -0.072 |
| C(a) | 50.05 | 46.22 | -3.83 | |
acetaldehyde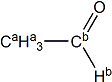 |
H(a) | 2.206 | 1.920 | -0.286 |
| H(b) | 9.789 | 9.573 | -0.216 | |
| C(a) | 30.89 | 28.35 | -2.54 | |
| C(b) | 199.93 | 184.61 | -15.32 | |
| cyclopropane |
H(a) | 0.198 | 0.236 | +0.038 |
| C(a) | -4.00 | -2.57 | +1.43 | |
propene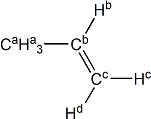 |
H(a) | 1.713 | 1.643 | -0.070 |
| H(b) | 5.734 | 6.004 | +0.270 | |
| H(c) | 4.963 | 5.014 | +0.051 | |
| H(d) | 4.883 | 5.218 | +0.335 | |
| C(a) | 17.90 | 18.96 | +1.06 | |
| C(b) | 139.00 | 129.43 | -9.57 | |
| C(c) | 114.39 | 113.52 | -0.87 | |
| propyne |
H(a) | 1.740 | 1.547 | -0.193 |
| H(b) | 1.936 | 1.928 | -0.008 | |
| C(a) | 3.33 | 3.90 | +0.57 | |
| C(b) | 85.96 | 71.36 | -14.20 | |
| C(c) | 71.94 | 66.95 | -4.99 | |
toluene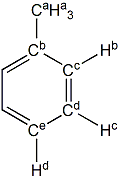 |
H(a) | 2.318 | 2.300 | -0.018 |
| H(b) | 7.061 | 7.406 | +0.345 | |
| H(c) | 7.138 | 7.523 | +0.385 | |
| H(d) | 7.042 | 7.361 | +0.319 | |
| C(a) | 21.41 | 20.48 | -0.93 | |
| C(b) | 137.83 | 134.97 | -2.86 | |
| C(c) | 129.09 | 126.15 | -2.94 | |
| C(d) | 128.28 | 126.75 | -1.53 | |
| C(e) | 125.38 | 122.93 | -2.45 | |
furan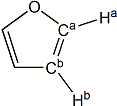 |
H(a) | 7.435 | 7.445 | +0.010 |
| H(b) | 6.380 | 6.252 | -0.128 | |
| C(a) | 142.64 | 136.81 | -5.83 | |
| C(b) | 109.54 | 105.73 | -3.81 | |
pyridine |
H(a) | 8.593 | 8.984 | +0.391 |
| H(b) | 7.231 | 7.242 | +0.011 | |
| H(c) | 7.617 | 7.965 | +0.348 | |
| C(a) | 149.94 | 148.58 | -1.36 | |
| C(b) | 123.75 | 118.00 | -5.75 | |
| C(c) | 135.89 | 136.32 | +0.43 | |
norbornene |
H(a) | 2.844 | 2.456 | -0.388 |
| H(b) | 5.989 | 6.340 | +0.351 | |
| H(c) | 0.950 | 0.988 | +0.038 | |
| H(d) | 1.610 | 1.455 | -0.155 | |
| H(e) | 1.076 | 1.010 | -0.066 | |
| H(f) | 1.313 | 1.327 | +0.014 | |
| C(a) | 41.84 | 36.82 | -5.02 | |
| C(b) | 135.36 | 133.91 | -1.45 | |
| C(c) | 24.67 | 23.61 | -1.06 | |
| C(d) | 48.60 | 44.56 | -4.04 | |
| HΔmax+ | +0.391 | |||
| HΔmax- | -0.388 | |||
| HΔabsavg | 0.180 | |||
| CΔmax+ | +1.43 | |||
| CΔmax- | -15.32 | |||
| CΔabsavg | 3.91 | |||
かなり長いTableですが、見てみると「軽い」モデル化学の割りに精度が良いのではないでしょうか。最後の6行について説明しますと、HΔmax+が1H-NMR化学シフトの正の最大誤差,HΔmax-が同じく負の最大誤差,HΔabsavgが同じく平均絶対誤差(誤差の絶対値の平均値)です。炭素についても同じです。1H-NMRの化学シフトのレンジがおよそ12ppm、13C-NMRの化学シフトのレンジがおよそ220ppmですので、平均絶対誤差はレンジに対して1.5%(1H),1.8%(13C)であり、かなり良い一致を見せています。ちなみに、NMR化学シフトの予測ツールとして、ChemDrawに付属している平面構造式と加成則から導くプログラム(ChemNMR)がよく知られていますが、そのようなツールでは、ノルボルネンのように空間を通して電子的環境が影響している分子では当然高い精度を望むべくもありません。しかし、量子計算から導かれる化学シフトはこのような分子に対しても等しい精度で求められます。実際、架橋メチレン位の2つの水素の化学シフトは非常によい一致を示しています(ChemNMRでは二つの水素を区別することすらできません)。ただ、トルエンの芳香環の1H化学シフトがいずれも低磁場側に大きくずれているのは気にはなりますf(^^;;
今回は、計算時間の都合でRHF/6-31g(d)で構造最適化及び遮蔽定数計算をしましたが、本来、NMRの議論を行うに当たっては、B3LYP/6-31g(d)による構造最適化、RHF/6-311+g(2d,p)による遮蔽定数計算が推奨されています。その為、ここに示したデータはあくまで低レベルなモデル化学での参考値です。それでもこれだけ真に迫った値が計算されるのですから、大したものです。
Leave a Reply
コメントを投稿するにはログインしてください。