有機化学の世界においては、対象となる分子の多くは閉殻分子で、かつ反応機構も極性機構で捉えられています。しかし、その枠組みから外れる分子や反応機構も数多く知られています。今後、電子構造や反応機構が見直されていくものもあるでしょう。
計算化学において取り扱いの難しい化学種に「ビラジカル」があります。よく知られているビラジカルといえば酸素分子がありますが、他にも実際に観測されたものや理論的に考えられるものがいろいろあります。これらは1重項・3重項ともに閉殻状態の理論(RHF)では取り扱うことができません。1重項なのに閉殻じゃない、というと変な感じがしますが、実際に1重項ビラジカルは観測されていますし(酸素分子は基底状態が3重項ですが、1重項酸素も光反応等において重要な役割を果たしています)、最も長寿命の1重項ラジカルは日本で生み出されました(広島大・安倍研)。
1重項ビラジカルのような特殊な電子構造を扱うには、複数の電子配置を一緒に考慮する手法が必要です。多数の電子配置を参照する理論を「多参照理論」と呼びますが、ここではGAMESSを使って、そのような理論の一つ「CASSCF(完全活性空間多配置SCF)」の簡単な計算例をお示しします。
はじめに―単参照理論での最適化
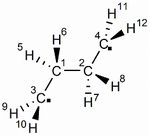
今回は、よく知られている直鎖形エチレン二量体(Fig.1)の1重項での安定構造と、二量化の遷移状態について計算を実施します。
まず、エチレン二量体の初期構造を作成します。この分子はD2hの対称性をもっており、Z-matrixで簡単に記述できます。私の記述例を以下に示します。
C C 1 rCC C 1 rCC 2 110 C 2 rCC 1 110 3 180 H 1 rCH 2 110 3 120 H 1 rCH 2 110 3 -120 H 2 rCH 1 110 4 120 H 2 rCH 1 110 4 -120 H 3 rCH 1 120 2 90 H 3 rCH 1 120 2 -90 H 4 rCH 2 120 1 90 H 4 rCH 2 120 1 -90 rCC=1.50 rCH=1.00
ナンバリングはFig.1の通りになっています。この構造を出発点に、計算を進めていくことにします。まず、単純にここから対称性を保ったままRHFやUHFで構造最適化するとどうなるでしょうか。
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE COORD=ZMT MAXIT=200 NZVAR=0 MULT=1 $END $SYSTEM TIMLIM=3600 MWORDS=1 $END $SCF DIRSCF=.T. DAMP=.T. $END $BASIS GBASIS=STO NGAUSS=3 $END $GUESS GUESS=HUCKEL $END $STATPT NSTEP=100 OPTTOL=0.0001 $END $DATA C4H8 biradical Cnh 2 C C 1 rCC C 1 rCC 2 110 C 2 rCC 1 110 3 180 H 1 rCH 2 110 3 120 H 1 rCH 2 110 3 -120 H 2 rCH 1 110 4 120 H 2 rCH 1 110 4 -120 H 3 rCH 1 120 2 90 H 3 rCH 1 120 2 -90 H 4 rCH 2 120 1 90 H 4 rCH 2 120 1 -90 rCC=1.50 rCH=1.00 $END
UHFの場合は$CONTRLのSCFTYPをRHF→UHFに変えるだけです。
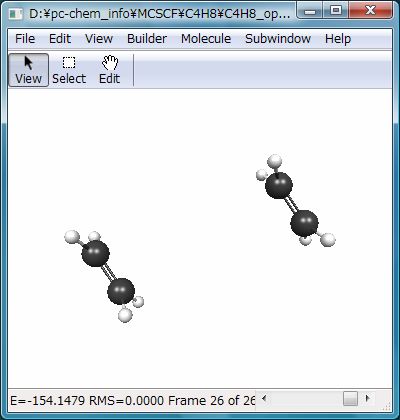
この計算は基底関数がSTO-3Gということもあり、すぐに終わります。結果を可視化ソフトで見ると…Fig.2のように、エチレン2分子に解離してしまいます。図はRHFの結果ですが、UHFでも同じ結果になります。二量体の構造は得られません。
では、CASSCFではどうでしょうか。
CASSCFの入力作成・構造最適化
CASSCFのような多配置SCFでは、計算成功の可否に初期軌道の質が大きく関わります。GAMESSでは、デフォルトで初期軌道をHuckel法により作成しますが、予めRHF等で収束した軌道を初期軌道に用いたほうが計算がスムーズに進行することが多いです。また、初期軌道をRHF等で作成する際に、同時にどの軌道を活性空間(AS)に取り込むかを調べます。
先のRHFによる構造最適化計算で、すでに初期構造の軌道は得られています(punchファイルの最初のほうに出力されています)が、軌道の可視化や$VECデータのコピーを容易にするため、もう一度1点計算をし直すことにします。

入力については省略しますが、計算により得られた分子軌道の中で大事なのは、今注目している部位に寄与の大きい軌道です。この例では、HOMO(16)とLUMO(17)(Fig.3)がラジカル電子の配置に大きく関わると考えられますので、この2軌道を活性空間に設定することにします。
CASSCFの入力例を以下に示します。
$CONTRL SCFTYP=MCSCF RUNTYP=OPTIMIZE COORD=ZMT MAXIT=200 NZVAR=0 MULT=1 $END $SYSTEM TIMLIM=3600 MWORDS=1 MEMDDI=1 $END $SCF DIRSCF=.T. DAMP=.T. $END $BASIS GBASIS=STO NGAUSS=3 $END $GUESS GUESS=MOREAD NORB=28 $END $MCSCF CISTEP=ALDET $END $DET NCORE=15 NACT=2 NELS=2 GROUP=C2h ISTSYM=1 NSTATE=1 WSTATE(1)=1 $END< $STATPT NSTEP=100 OPTTOL=0.0001 HSSEND=.T. $END $DATA C4H8 biradical Cnh 2 C C 1 rCC C 1 rCC 2 110 C 2 rCC 1 110 3 180 H 1 rCH 2 110 3 120 H 1 rCH 2 110 3 -120 H 2 rCH 1 110 4 120 H 2 rCH 1 110 4 -120 H 3 rCH 1 120 2 90 H 3 rCH 1 120 2 -90 H 4 rCH 2 120 1 90 H 4 rCH 2 120 1 -90 rCC=1.50 rCH=1.00 $END $VEC 1 1 7.00849008E-01 3.30950787E-02-2.49690729E-03-1.73512246E-03-0.00000000E+00 1 2-7.00849008E-01-3.30950787E-02-2.49690729E-03-1.73512246E-03-0.00000000E+00 1 3 2.24185518E-04-4.28773522E-03 2.81846799E-03-1.45679763E-03-0.00000000E+00 1 4-2.24185518E-04 4.28773522E-03 2.81846799E-03-1.45679763E-03-0.00000000E+00 1 5-5.80164897E-03 5.80164897E-03-5.80164897E-03 5.80164897E-03 3.26677990E-05 1 6-3.26677990E-05 3.26677990E-05-3.26677990E-05 2 1 7.01576251E-01 2.41583745E-02 2.08415794E-03 2.84541396E-03-0.00000000E+00 (中略) 27 6-6.27385743E-01-6.27385743E-01 6.27385743E-01 28 1-0.00000000E+00-0.00000000E+00-0.00000000E+00-0.00000000E+00 7.05445020E-01 28 2-0.00000000E+00-0.00000000E+00-0.00000000E+00-0.00000000E+00-7.05445020E-01 28 3-0.00000000E+00-0.00000000E+00-0.00000000E+00-0.00000000E+00-6.94066062E-01 28 4-0.00000000E+00-0.00000000E+00-0.00000000E+00-0.00000000E+00 6.94066062E-01 28 5 4.55138447E-01 4.55138447E-01-4.55138447E-01-4.55138447E-01-4.91636497E-01 28 6-4.91636497E-01 4.91636497E-01 4.91636497E-01 $END
まず$CONTRLでSCFTYP=MCSCFとします。これが無ければ始まりません。これに対応するのが$MCSCFで、ここでCIのやり方を選び(CISTEP=ALDET)、またSCFのオプションを選択したりします。CASSCFを実行するに当たって、一番直感的に入力を作成できるのがALDETですので(これは私の主観ですが)、ここではそれを選択しています。他にGUGAやORMASなどが選択でき、これらもよく使われます。
CISTEP=ALDETに対応するのが$DETで、ここで活性空間の設定をします。NCOREは励起配置に取り込まない被占軌道の数、NACTは活性軌道の数、NELSは活性軌道に配置する電子数です。これで、一般によく書かれる「CASSCF(2,2)」を指定したことになります。GROUPは点群で、通常は$DATAグループから引用されますが、$DETで指定できるのは点群の中でもアーベル群(C1, Ci, Cs, C2, C2v, C2h, D2, D2h)だけです。ISTSYMは解く状態の対称性を指定します。指定は数字ですが、これはマニュアルに従います。上記の例はC2hですが、この場合1=Ag, 2=Bu, 3=Bg, 4=Auとなり、基底状態の計算ですので1を指定します(励起状態ではそれぞれに応じた数字を指定します)。
NSTATEは解く電子状態の数、WSTATEは各電子状態の重み付けを表す配列です(ここでは1つの状態しか解かないのでこのようになっていますが、励起状態の計算等で複数の電子状態を解いてその内下から2番目の状態を使うような場合は、NSTATE=5 WSTATE(1)=0,1,0,0,0のように記述したりします)。
$GUESSでGUESS=MOREADとし、初期軌道として$VECを指定します。NORBで$VECに含まれる軌道数($VECの行頭の数字、事前に軌道を確認する際、全軌道数も同時にチェックするとよい)を指定します。$VECは、RHF一点計算のpunchファイルからコピーします(PC GAMESSのオフィシャルサイトで配布されている入出力ファイル処理インタプリタ「JOB」を使うと作業を簡単にできます)。 また、$SYSTEMでMEMDDIを指定していますが、MCSCFで解析的にHessianを計算する場合はMEMDDIが必要になります(マニュアルを参照して下さい)。
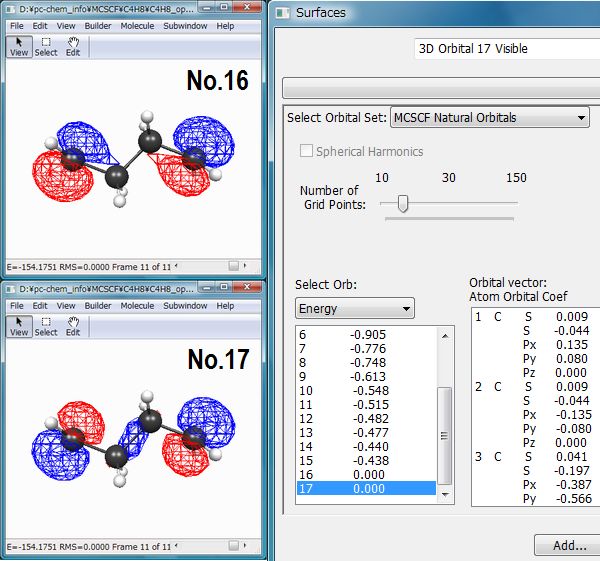
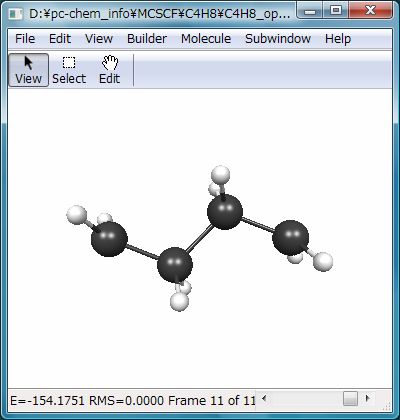
この計算の結果、Fig.4のような構造が得られます。最適化されたMCSCF自然軌道を可視化すると、二つの電子がほぼ一つずつ同じエネルギーレベルの非結合性軌道(縮退した軌道)に入っていることがわかります(Fig.5)。振動解析からも、これが停留点であることが確認できます。出力ファイルには、電子配置と最適化されたCI係数も出力されています(下記)。
CI EIGENVECTORS WILL BE LABELED IN GROUP=C2H PRINTING ALL NON-ZERO CI COEFFICIENTS STATE 1 ENERGY= -154.1751291596 S= 0.00 SZ= 0.00 SPACE SYM=AG ALPH|BETA| COEFFICIENT ----|----|------------ 10 | 10 | 0.7672155 01 | 01 | -0.6413894
遷移状態の最適化とIRC計算
無事1重項のエチレン二量体の構造を得て、その電子構造も把握することができました。エチレン2分子からこの構造は生成すると考えられますから、次にその中間の構造…遷移状態を探索することにします。
上記計算で最適化された構造のZ-matrixの中で、中央のC-C結合を2.0Åに変更し、再度RHFの一点計算を実施します。
C C 1 2.0000000 ←1.5652118から変更 C 1 1.5134808 2 112.2042627 C 2 1.5134808 1 112.2042627 3 180.0000000 0 H 1 1.0886017 2 108.7235173 3 121.6016116 0 H 1 1.0886017 2 108.7235173 3 -121.6016116 0 H 2 1.0886017 1 108.7235173 4 121.6016116 0 H 2 1.0886017 1 108.7235173 4 -121.6016116 0 H 3 1.0836564 1 117.6191033 2 72.2417168 0 H 3 1.0836564 1 117.6191033 2 -72.2417168 0 H 4 1.0836564 2 117.6191033 1 72.2417168 0 H 4 1.0836564 2 117.6191033 1 -72.2417168 0
得られた初期軌道(punch内の$VEC)と上記Z-matrixを使って、遷移状態探索の入力を作成します。このケースでは、結合長が伸びたものの活性空間に取り込むべき軌道はHOMOとLUMOで変わっていません。
$CONTRL SCFTYP=MCSCF RUNTYP=SADPOINT COORD=ZMT MAXIT=200 NZVAR=0 MULT=1 $END $SYSTEM TIMLIM=3600 MWORDS=1 MEMDDI=1 $END $SCF DIRSCF=.T. DAMP=.T. $END $BASIS GBASIS=STO NGAUSS=3 $END $GUESS GUESS=MOREAD NORB=28 $END $MCSCF CISTEP=ALDET $END $DET NCORE=15 NACT=2 NELS=2 GROUP=C2h ISTSYM=1 NSTATE=1 WSTATE(1)=1 $END $STATPT NSTEP=100 OPTTOL=0.0001 HESS=CALC HSSEND=.T. $END $DATA C4H8 biradical Cnh 2 C C 1 2.0000000 C 1 1.5134808 2 112.2042627 C 2 1.5134808 1 112.2042627 3 180.0000000 0 H 1 1.0886017 2 108.7235173 3 121.6016116 0 H 1 1.0886017 2 108.7235173 3 -121.6016116 0 H 2 1.0886017 1 108.7235173 4 121.6016116 0 H 2 1.0886017 1 108.7235173 4 -121.6016116 0 H 3 1.0836564 1 117.6191033 2 72.2417168 0 H 3 1.0836564 1 117.6191033 2 -72.2417168 0 H 4 1.0836564 2 117.6191033 1 72.2417168 0 H 4 1.0836564 2 117.6191033 1 -72.2417168 0 $END $VEC 1 1 7.01393784E-01 2.63548842E-02 1.03921524E-04 4.85742852E-04 0.00000000E+00 1 2-7.01393784E-01-2.63548842E-02 1.03921524E-04 4.85742852E-04 0.00000000E+00 1 3-4.94414687E-04-4.06587222E-03 2.62667863E-03-1.54070630E-03 0.00000000E+00 (以下略)
$CONTRLでRUNTYP=SADPOINTとし、$STATPTでHESS=CALCとしています(小さな分子であれば、HESS=CALCでも時間はかかりませんが、大きい分子でこれをやると非常に時間がかかるため、その場合は別途低いレベルのモデル化学でHessian計算をしてHESS=READとした方が賢明です)。
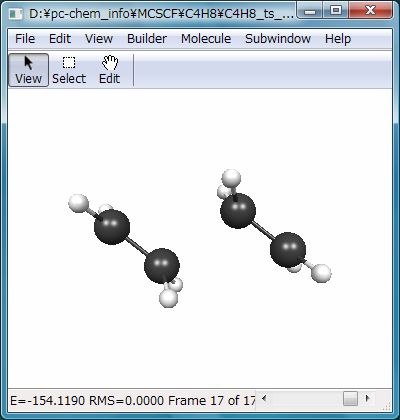
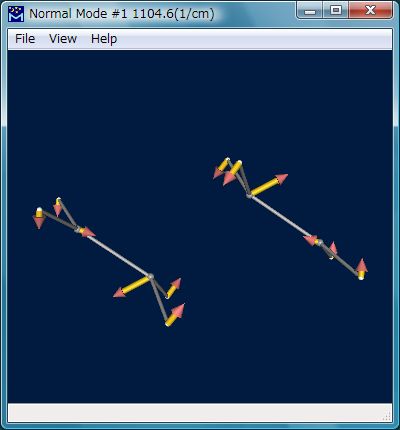
この計算の結果、Fig.6のような構造が遷移状態として得られます。この構造はFig.7のような唯一つの虚振動モード(1104.58i cm-1)を有し、意図した反応の遷移状態であることが裏付けられます。
遷移状態が得られましたので、そこから原系・生成系に向かうIRCを辿ることができます。この計算で静的な反応経路がはっきりとしたものになります。入力は例えば以下のようになります。
$CONTRL SCFTYP=MCSCF RUNTYP=IRC COORD=ZMT MAXIT=200 NZVAR=0 MULT=1 $END $SYSTEM TIMLIM=3600 MWORDS=1 $END $SCF DIRSCF=.T. DAMP=.T. $END $BASIS GBASIS=STO NGAUSS=3 $END $GUESS GUESS=MOREAD NORB=28 $END $MCSCF CISTEP=ALDET $END $DET NCORE=15 NACT=2 NELS=2 GROUP=C2h ISTSYM=1 NSTATE=1 WSTATE(1)=1 $END $IRC SADDLE=.T. NPOINT=5 FORWRD=.T. $END $DATA C4H8 biradical Cnh 2 C C 1 2.0356362 C 1 1.3676055 2 106.1469352 C 2 1.3676055 1 106.1469352 3 180.0000000 0 H 1 1.0826655 2 96.7838113 3 122.3220097 0 H 1 1.0826655 2 96.7838113 3 -122.3220097 0 H 2 1.0826655 1 96.7838113 4 122.3220097 0 H 2 1.0826655 1 96.7838113 4 -122.3220097 0 H 3 1.0802514 1 121.4087570 2 84.7038773 0 H 3 1.0802514 1 121.4087570 2 -84.7038773 0 H 4 1.0802514 2 121.4087570 1 84.7038773 0 H 4 1.0802514 2 121.4087570 1 -84.7038773 0 $END $VEC 1 1 7.01393784E-01 2.63548842E-02 1.03921524E-04 4.85742852E-04 0.00000000E+00 1 2-7.01393784E-01-2.63548842E-02 1.03921524E-04 4.85742852E-04 0.00000000E+00 1 3-4.94414687E-04-4.06587222E-03 2.62667863E-03-1.54070630E-03 0.00000000E+00 (中略) 28 4 0.00000000E+00 0.00000000E+00 0.00000000E+00 0.00000000E+00-6.67420878E-01 28 5-3.76447975E-01-3.76447975E-01 3.76447975E-01 3.76447975E-01 4.59743371E-01 28 6 4.59743371E-01-4.59743371E-01-4.59743371E-01 $END $HESS ENERGY IS -154.1190309087 E(NUC) IS 113.2868050473 1 1 4.99381791E-01-6.16530232E-01 0.00000000E+00 7.73159893E-01-1.08030177E-01 1 2 0.00000000E+00-5.05503920E-01 3.35419782E-01 0.00000000E+00-5.25312149E-01 1 3 2.78099599E-01 0.00000000E+00-7.41763169E-02 7.05381343E-02-7.68580025E-02 (中略) 36 6 5.87569201E-05 1.79303061E-04-1.43812534E-02 1.63040204E-02-1.26706340E-02 36 7-8.87988193E-05 3.95914479E-05-1.84563136E-04 1.28008125E-01-1.36162559E-01 36 8 3.75942986E-01 $END
$CONTRLでRUNTYP=IRCとし、これに対応する$IRCを記述します。SADDLEは、入力構造が遷移状態であることを示し、NPOINTは何点までIRC上を進むかです(その遷移状態が正しく原系と生成系を結んでいるかを確かめるためなら、多くはいらないはず)。FORWRDは$HESSの虚振動モードにしたがって変位させるか(true)、その逆に変位させるか(false)で、ここだけが異なる2つの入力ファイルを作成して実行することで、遷移状態の両サイドへIRCを辿ることができます。$HESSは先の遷移状態最適化計算のpunchファイルよりコピーします。
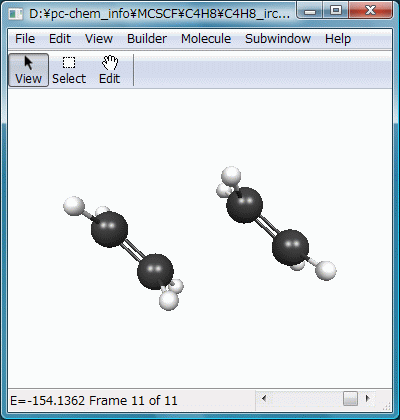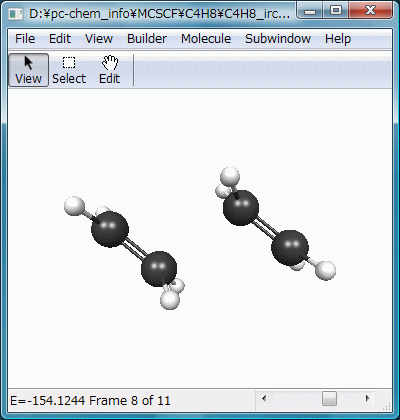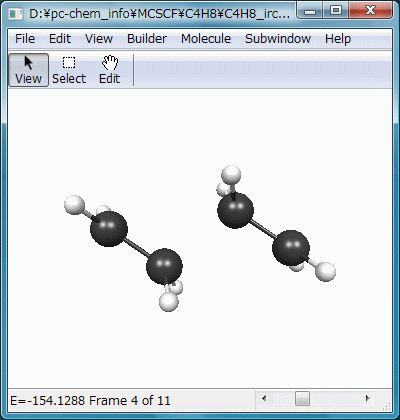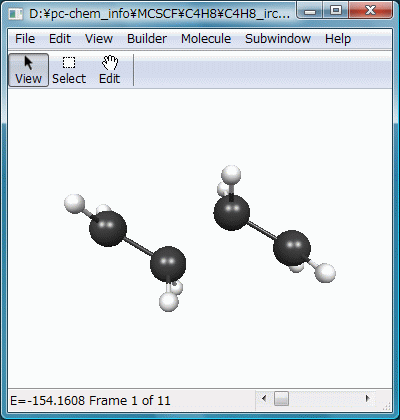
結果をアニメーション化したのがFig.8で、2分子のエチレンが接近し、遷移状態を通過してビラジカルを生成する様子が滑らかに再現されています。
注) 本稿で実施した計算はシンプルなモデル化学(CASSCF(2,2)/STO-3G)で実施していますので、定量性は期待できません。ビラジカルの構造も基底関数の改良で変化しますが(特にラジカル周りの平面性など)、ここではその手の話が本筋ではないので割愛しています。
[本稿で行った計算の入出力ファイル(618 KB)]
Leave a Reply
コメントを投稿するにはログインしてください。