CCL (computational chemistry list)で無償公開されているMOPAC 6のちょっと強化版「MOPAC 6.06」をここに公開します。
mopac606_exe.zip
(Windows用実行ファイル(Cygwin/g77でコンパイル), 重原子80/水素原子100)
mopac606_src_diff.zip
(改変・追加した差分ソースコード)
linux_runscript.zip
(linux用入出力補助シェルスクリプト)
MOPAC 6.06とは
Yale大学のJorgensenらによるPDDG (Pairwise Distance Directed Gaussian)法追加版MOPAC 6を基に、同グループから発表された追加のPDDG/PM3パラメータを加え、いくつかの機能追加・バグフィックスを施したのがMOPAC 6.06です。発案およびPDDG/PM3パラメータ追加はs2k(pc-chem.info)が、.mgf出力やいくつかのバグ修正等は千田(tencube)が実施しました。
MOPAC 6とMOPAC 6.06の主な違い
・既報の全PDDG/PM3パラメータを収載(H, C, N, O, F, Si, P, S, Cl, Br, I)
※PDDG/MNDOはオリジナルソースコードに記述済みのC, H, N, Oのみ
| 分子セット | MAE of HOF (kcal/mol) | |||
|---|---|---|---|---|
| AM1 | PM3 | PDDG/PM3 | ||
| CHNO | 6.7 | 4.4 | 3.2 | |
| halogens | 11.1 | 8.1 | 5.6 | |
| Si | 11.7 | 11.1 | 11.9 | |
| P | 18.2 | 21.5 | 17.9 | |
| S | 10.6 | 10.5 | 6.4 | |
| All | 10.8 | 8.7 | 6.5 | |
 ・Jmol等で可視化できるFormatted Graphic File (.mgf)の出力が可能
・Jmol等で可視化できるFormatted Graphic File (.mgf)の出力が可能
・.arcファイルの電荷の出力バグを修正
公開・利用・再配布条件
・元プログラムに従い、PDS (Public Domain Software)と同様の条件に従うものとします。
(現行の日本国内の法律では著作権を放棄できませんが、自由な改変・配布を認めます)
・改変したものを公開する際は、ソースコードも公開して下さい。
参考文献・URL
○オリジナルソースコード
・MOPAC 6 (http://server.ccl.net/cca/software/SOURCES/FORTRAN/mopac6_sources/)
・PDDG (http://zarbi.chem.yale.edu/doc/pddg/)
※PDDGのソースコードの利用については原著者に連絡の上、了承済みです。
○文献
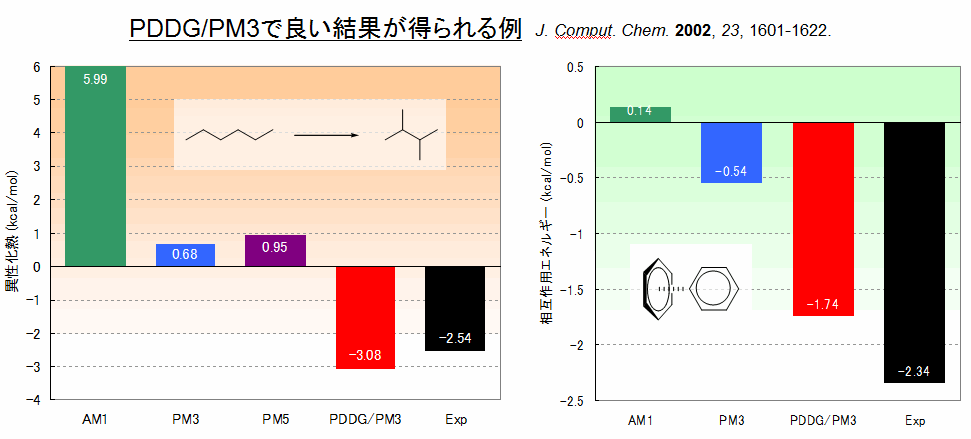
・PDDG (C, H, N, O): J. Comput. Chem. 2002, 23, 1601-1622.
・PDDG (Si, P, S): J. Chem. Theory Comput. 2005, 1, 817-823.
・PDDG (F, Cl, Br, I): J. Comput. Chem. 2004, 25, 138-150.
簡易マニュアル
(1)コンパイル方法
1-0. Windowsならcygwinを導入し、g77が使えるのが前提。Linuxでもg77が使えるのが前提。
1-1. ここからMOPAC 6のソースコードをダウンロードし、任意のフォルダに展開する。
1-2. ここからPDDG差分ソースコードをダウンロードし、先のフォルダに上書きする。
1-3. mopac606_src_diff.zipをダウンロードし、先のフォルダに上書きする。
1-4. コマンドライン(cygwin/bashかLinuxの端末)でmakeを実行。
1-5. フォルダ内にmopac606.exeが生成する。
※「SIZES」というファイル内のMAXHEV, MAXLITの値を変更することで、扱える原子数を変更できます
(2)計算の実行方法
2-a. 実行ファイルと同じフォルダ内に入力ファイルを「FOR005」というファイル名で作成し、実行ファイルを起動する。
2-b. Windows上でWinmostarから利用可能。適当なフォルダに実行ファイルを入れ、Winmostarのメニュー[その他]→[パスの設定]で指定する。
2-c. Linux上での入出力処理にはこちらのシェルスクリプト(bashで記述)も利用可能。
(3)追加機能のキーワード
3-a. PDDG/PM3で計算を行うには「PDG」、PDDG/MNDOで計算を行うには「MDG」を指定。
3-b. Formatted Graphic Fileは「GRAPH」「GRAPHF」どちらのキーワードでも出力される。
NBO 3.0の統合
[注意] NBO 3.0は、すでにサポート外となっている古いプログラムです。バグがいくつか残されていることも明らかになっていますので、利用は自己責任でお願いします。原著者のF. Weinhold教授への問い合わせは絶対に避けることと、サポートを受けたい場合は最新版のNBO6を購入されることをお勧めします。
CCLに、現在広く使われている密度解析プログラム「NBO (Natural Bond Orbital)」の旧版(ver.3.0)が公開されています。
http://server.ccl.net/cca/software/MS-WIN95-NT/mopac6/
このソースコードを、MOPAC 6.06に統合することができます。
1. 上記URLからNBO 3.0のソースコード(Executable with NBO & Sources)をダウンロードし、任意のフォルダに展開する。
2. mopnbo.fの5872行目「DIMENSION BASIS(4)」を「CHARACTER*4 BASIS(4)」に変更し、「NBREAD」と「NBWRIT」サブルーチンをそれぞれ「nbread.f」「nbwrit.f」として別ファイルに切り離す。
3. MOPAC 6.06のソースコードにmopnbo.f, nbread.f, nbwrit.fを追加する。(writenbo.fは必要ない)
4. writmo.fの502行目のコメントマーク(行先頭の”C”)を半角スペースに置換し「CALL RUNNBO」を有効化。
5. Makefileの変数OBJの末尾(「xyzint.o \」の後ろ)に「mopnbo.o \」「nbread.o \」「nbwrit.o」の3行を追加。
6. makeを実行。
NBO解析を行うには、座標指定の後に一行空けて「$NBO $END」。$NBOと$ENDの間にキーワードを挿入してオプション指定が可能。(詳しくはこちらのマニュアルを参照)
Leave a Reply
コメントを投稿するにはログインしてください。