有機合成化学は、Vaska型錯体に代表される有機遷移金属錯体の発見から数十年の時を経て、ようやくその恩恵にあずかることができるようになりました。有機金属とは、炭素-金属結合を有する化合物の総称で、特に、遷移金属との錯体を有機遷移金属錯体と称します。有機金属は通常の有機化学反応では起こり得ない数々の反応を生み出し、有機合成の方法論に新たな道筋を付けることになりました。均一系水素化,クロスカップリング,オレフィンメタセシス…これらは現在最も利用されている遷移金属触媒反応の一例ですが、いずれも通常の有機化学では不可能な反応でありながら高い収率・選択性を達成することができます。
えー、自分の専門であるあまり小難しいことを列挙しましたが、要は、遷移金属は炭素原子と (通常の有機化学の視点からは)不可解な錯体を驚くほど安定に生成し、そしてそれらは驚くべき反応性を示すのです。ここでは、そんな有機遷移金属錯体をモデリングしつつ、紹介していきたいと思います。
フェロセン
まず最初に、世界に衝撃を与えたサンドイッチ型有機金属錯体、フェロセンを紹介したいと思います。フェロセンは、鉄イオンがシクロペンタジエニルアニオン(Cp)に上下からサンドされた独特の構造を有しています。この化合物を最初に合成した方は、この構造に思い至らず間違った構造を提唱しましたが、Woodwardらがその構造に疑問を抱き自ら合成、当時の最新鋭の技術・NMRによりサンドイッチ型構造を実証したのは余りにも有名です。
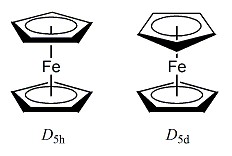
フェロセンには実は2種類の構造があります。上下2つのCp環が同じ向きに重なった「D5h」対称の配座と、互い違いに重なった「D5d」対称の配座です(Fig.1)。実験的には気相中ではD5h、結晶ではD5dの構造をとっています。そこで、PC GAMESSで真空中での構造最適化を行ってみましょう。気相中の構造を再現すると考えられますので、D5hの対称性で計算を実施してみます。

対称性が非常に高い分子の計算を行う場合、通常はモデリングソフトを使うよりも自分の手で入力を作成した方が早いことが多いです。t例えばフェロセンの単位構造(GAMESSでいう”Unique Atoms”)はFe,C,Hそれぞれ1個からなる非常に単純な構造です(Fig.2)。この単位構造と対称性を与えれば、プログラムが自動的に残りの原子を配置してくれます。
D5h対称のフェロセンを、RB3LYP/631dLAN(混合基底系:6-31G(d) for C and H, LANL2DZ for Fe)で構造最適化し、続けて振動計算を行う(GaussianでいうOpt Freq)入力ファイルの例を以下に示します。
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE COORD=UNIQUE
MAXIT=200 NZVAR=3 ICHARG=0 MULT=1 DFTTYP=B3LYP
ECP=READ $END
$SYSTEM TIMLIM=600 MWORDS=2 AOINTS=DIST
L2SIZE=1024 VOLSIZ=512 $END
$STATPT NSTEP=500 OPTTOL=0.00001 HSSEND=.T. $END
$BASIS EXTFIL=.T. GBASIS=631dLAN $END
$SCF DIRSCF=.T. DAMP=.T. FDIFF=.F. $END
$ZMAT DLC=.T. AUTO=.T. $END
$GUESS GUESS=HUCKEL $END
$DATA
Ferrocene,D5h
Dnh 5
Fe 26.0 0.000000 0.000000 0.000000
C 6.0 1.200000 0.000000 1.660000
H 1.0 2.200000 0.000000 1.660000
$END
$ECP
FE-ECP GEN 10 2
3 ----- d potential -----
-10.00000000 1 392.61497870
-63.26675180 2 71.17569790
-10.96133380 2 17.73202810
5 ----- s-d potential -----
3.00000000 0 126.05718950
18.17291370 1 138.12642510
339.12311640 2 54.20988580
317.10680120 2 9.28379660
-207.34216490 2 8.62890820
5 ----- p-d potential -----
5.00000000 0 83.17594900
5.95359300 1 106.05599380
294.26655270 2 42.82849370
154.42446350 2 8.77018050
-95.31642490 2 8.03978180
C-ECP NONE
C-ECP
C-ECP
C-ECP
C-ECP
C-ECP
C-ECP
C-ECP
C-ECP
C-ECP
H-ECP NONE
H-ECP
H-ECP
H-ECP
H-ECP
H-ECP
H-ECP
H-ECP
H-ECP
H-ECP
$END
この入力ファイルには基本的な入力以外に3点重要な点があります(1.点群を指定した入力 2.ECPの入力 3.外部基底関数の利用)がありますが、それらの解説は別の記事に譲ります。同様にして、MP2やHFでの入力も作成、実行してみましょう。
気相でのフェロセンのFe-Cp結合距離(FeとCp環の中心との距離)は1.66Åです。tableにいくつかの理論モデル(基底関数はいずれも631dLAN)で計算したFe-Cp結合距離を示します。 RHFでは結合距離はかなり長めに算出され、逆にRMP2では短めに算出されます。それに対し、hybrid-DFT(RB3LP,RPBE0)では実測値と非常に良い一致を示しています。
| 理論モデル | Fe-Cp結合距離(Å) |
| 実験値 | 1.66 |
| RHF | 1.875 |
| RMP2 | 1.530 |
| RB3LYP | 1.686 |
| RPBE0 | 1.645 |
カルボニル錯体
フェロセンよりももっと昔から知られている重要な有機金属錯体に、金属カルボニルがあります。その中でも早くに見出されたニッケルカルボニルNi(CO)4は、メタンと同じTd対称を有する錯体で、猛毒の揮発性液体でもあります。カルボニル(一酸化炭素)錯体は、周期表の属に従って配位数と構造(対称性)を変化させます。以下に、いくつかのカルボニル錯体の最適化構造を示します。
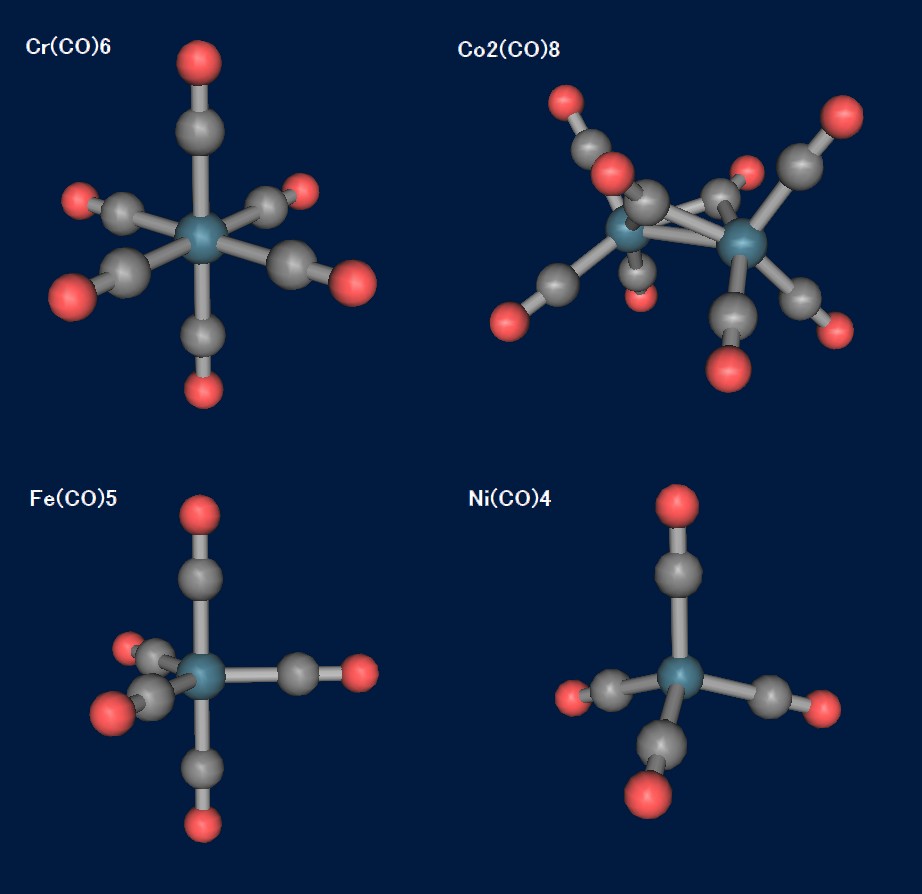
いずれも重要な錯体で、Cr,Co,Feの錯体は有機合成にも応用されています(Co錯体はPauson-Khand反応で有名ですね…Niはその毒性の強さのため、初期の研究以降は殆ど利用されていません)。各構造はRB3LYP/DZVPで最適化しました(いずれも対称性を利用)。基底関数DZVPは、ESML Basis Set Exchangeより入手したものを外部基底ファイル化して利用しています。
メタラベンゼン
遷移金属錯体は今のところ、新規反応開拓と有機合成への応用を中心として研究が進められているように思いますが、単純にその構造が面白いと思えるものも中にはあります。Metallabenzeneとは環の構成要素として金属原子を含むベンゼンで、「そんなものできるの?」と思いたくなりますが、様々な遷移金属元素で合成されています。ここでは例としてIridabenzeneとPlatinabenzeneを紹介します。Iridabenzeneの構造最適化では、入力ファイルこのような形になります。
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE COORD=UNIQUE EXETYP=RUN MAXIT=200 NZVAR=27 ICHARG=0 MULT=1 DFTTYP=PBE0 ECP=SBKJC $END $SYSTEM TIMLIM=600 MWORDS=2 AOINTS=DIST L2SIZE=1024 VOLSIZ=512 $END $STATPT NSTEP=500 OPTTOL=5E-5 DXMAX=0.2 $END $BASIS GBASIS=SBKJC NDFUNC=1 $END $SCF DIRSCF=.T. DAMP=.T. $END $ZMAT DLC=.T. AUTO=.T. $END $GUESS GUESS=HUCKEL $END $DATA Iridabenzene RPBE0/SBKJC-d Cnv 2 Ir 77 0 0 0 P 15 0 2.31 -0.36 C 6 0 0 3.4 C 6 1.24 0 2.76 C 6 1.4 0 1.37 H 1 0 0 4.48 H 1 2.41 0 1 H 1 2.13 0 3.38 H 1 0 2.6 -1.75 H 1 1.1 3.06 0.1 Cl 17 1.83 0 -1.71 $END
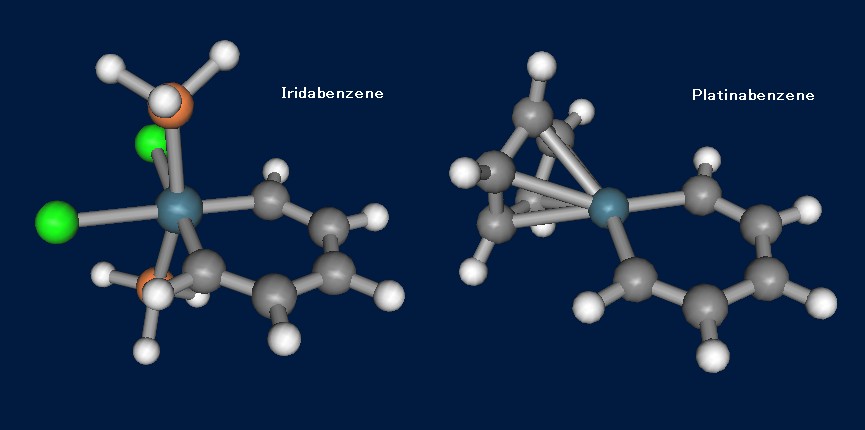
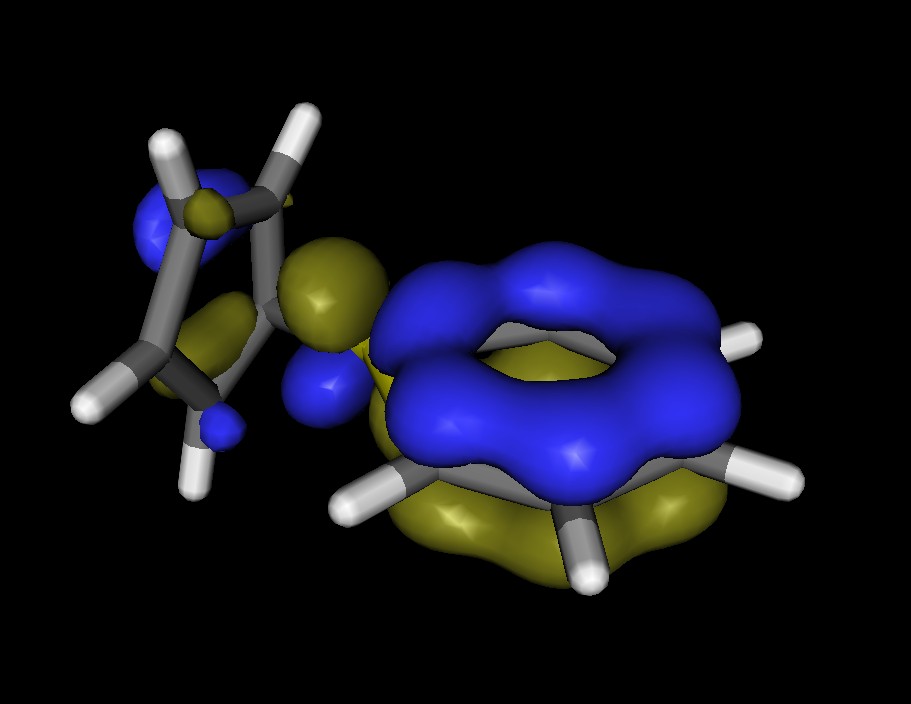
この入力では、GAMESSの実行ファイル内に組み込まれているECP基底SBKJCを利用し、C2v対称を指定しています。最適化されたIridabenzeneとPlatinabenzeneの構造をFig.4に示します。どちらも金属原子に配位子が乗っているので、果たしてπ共役があるのかが見た目ではよく分かりません。Fig.5は、Platinabenzeneの”ベンゼン環”の軌道(HOMO-10)をMolekel 5.1で可視化したものですが、Ptのd軌道が関与していて、炭素だけのベンゼン環のπ軌道と見た目がほとんど変わらないことがよくわかると思います。
[本稿で行った計算の入出力ファイル(2.16 MB)]
Leave a Reply
コメントを投稿するにはログインしてください。