高校の化学で習う有機化合物の分類に「芳香族化合物」があります。由来を知らないと何か変なネーミングですが、変なのはそれだけではなく、むしろその化学的性質です。不飽和結合(=π結合)を有するにも関わらず付加反応を受けにくく、むしろ置換反応を起こす。普通の不飽和化合物としての予測よりも燃焼熱が小さい。ベンゼンやシクロペンタジエニルアニオンのC-C結合が全て同じ長さ。こういった芳香族化合物に特徴的な性質を「芳香族性」といいます。 計算化学によって芳香族性を調べることができますが、ここでは
- 芳香族性による安定化を実験的に見積もる方法である水素化熱の比較
- 異性化反応モデルによる芳香族性の見積もり
- 磁気的性質によって芳香族性・反芳香族性を調べる
の3つについて計算例をお示しします。
水素化熱の計算
有機化学の教科書の芳香族化合物の章には必ず載っている、水素化熱の比較グラフ。これを計算で再現してみます。
まず、シクロヘキサン(D3d)・シクロヘキセン(C2)・1,3-シクロヘキサジエン(C2)・ベンゼン(D6h)の構造最適化と振動解析行います(それぞれ分子の対称性に気をつけて入力を作成)。なお、実測値は25 ℃ (=298.15 K), 1 atmでのエンタルピー変化ですので、計算も同条件で実施し比較する必要があります。今回の計算例はMP2/6-31G(d)で行いました。
計算結果をFig.1にまとめました。シクロヘキサンとシクロヘキセン+H2、シクロヘキサンと1,3-シクロヘキサジエン+2H2を比較すると、前者の2倍よりも後者の方が2.55 kcal/molだけ小さいことがわかります。この差は、二重結合が隣接して並んだこと(共役)による安定化エネルギー(dHconjugate)です。シクロヘキサンからシクロヘキセンへの脱水素エネルギーが二重結合を導入するのに必要なエネルギー(dHC=C, 計算値28.95 kcal/mol)だとすると、仮想ベンゼン(1,3,5-シクロヘキサトリエン)は二重結合が3つあり、共役も3箇所ありますから、3 × dHC=C – 3 × dHconjugate = 79.21 (kcal/mol)となります。実際のベンゼンは 43.41 kcal/molで、この2つの差 35.80 kcal/mol が「芳香族性獲得による安定化エネルギー (dHarom)」であると考えられます。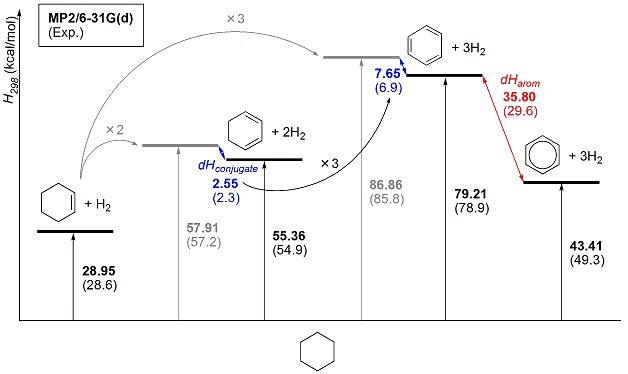
並べて実測値※1も記載してあります(括弧内の値)。シクロヘキセン、シクロヘキサジエンの計算値は実測に非常によく一致する一方、ベンゼンの水素化熱はやや誤差が残る結果となりました。
異性化反応を使った芳香族性の推算
芳香族性によって獲得する安定化エネルギーを見積もる方法として、ISE(Isomerization Stabilization Energy)がSchleyerらによって提唱されています(2002年)※2。これは、Eq.1のような反応を考えます。環状共役系の中の1つの二重結合がexo-メチレンとendo-メチレンに相互変換する時のエネルギー差(を補正したもの)が、共役系が環状になったことによる安定化となり、芳香環ならば芳香族性獲得による安定化ということになるわけです。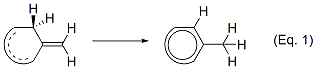
含酸素芳香環の代表例・フランで、ISEの算出をしてみます。
フランでEq.1と同様な式を立てるとEq.2のようになります。先のISEの説明で「を補正したもの」という文言がありましたが、それは環状共役とは独立した「exo-endo間の異性化エネルギー」の補正です。フランの場合、環ひずみも考慮してEq.3のような補正式を立てます。Eq.2の異性化エンタルピーにEq.3の異性化エンタルピーを加えることで、補正ISE(ISEcorrected)とします。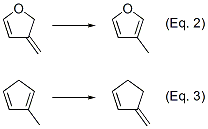
計算は水素化熱の算出と同様で、構造最適化・振動解析を行います。ここではより計算精度を高めるため、全電子エネルギーEelだけ、より精度の高いモデル化学で再計算しています(具体的にはPBE0/pc-2//PBE0/6-31G(d))。結果として、Eq.2の異性化エンタルピーは -16.43 kcal/mol, Eq.3の異性化エンタルピーは +3.55 kcal/molとなり、ISEcorrectedは -12.87 kcal/molと算出されました。ホモデスミック反応(Eq.4)のdH(G2MP2)が-12.2 kcal/molと報告されていますが※3、それに近い値になっています。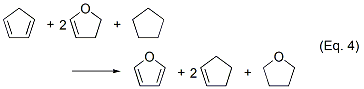
NICS:磁気的性質による芳香族性の定量化
ISEを提案したSchleyerは、1996年に分子の磁気的性質…環の中心の化学シフトが芳香族性の尺度になるとして、NICS(Nucleus-Independent Chemical Shift)を提案しています※4。これは環が芳香族ならその環電流効果によって環の中心が強く遮蔽化され、反芳香族なら逆に反遮蔽化されることに由来します。NICSは、NMR計算と全く同じ手順で行い、環の中心に数学的な点(電荷も軌道もない純粋な点)を配置し、その点での化学シフトの計算値の正負を入れ替えた値になります。ここで言う「環の中心」とは、環を構成する原子の座標の平均値です(重心ではありません)。
一般に、環の中心のNICSを「NICS(0)」と表記し、環の面に対して1 Åだけ浮いた座標のNICSを「NICS(1)」と表記します。平面の芳香環では、同じNICSでも環の中心よりもすこし浮いたところの方が、周囲のσ結合の影響を最小限に留めたより純粋なπ電子による効果を反映します(そのため、3員環のNICS(0)はσ結合による遮蔽効果が大きく芳香族性の評価に使えません)。
GAMESSでの具体的な入力例を以下に示します。原子ラベルは何でもいいですが、Gaussianでゴースト原子を意味する「Bq」をこの例では使っています。陽子数は0とし(これによってGAMESSはゴースト原子と判別します)、あとは環の中心となる座標(またはそこから1 Å浮いた座標)を指定します。他は通常のNMR計算と変わりません。ゴースト原子の指定は分子の後ろで指定した方が良いです(前に置くとSCF後の密度解析の出力がおかしくなる)。
$CONTRL SCFTYP=RHF RUNTYP=NMR COORD=UNIQUE MAXIT=200 NZVAR=0 $END $SYSTEM TIMLIM=36000 MWORDS=100 $END $SCF DIRSCF=.F. DAMP=.T. $END $BASIS GBASIS=N311 NGAUSS=6 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $DATA Borole, NMR, RHF/6-311G(d) C1 B 5.0 0.0000000000 0.0000000000 -1.3279447109 C 6.0 -1.2526668203 0.0000000000 -0.3676675132 C 6.0 1.2526668203 0.0000000000 -0.3676675132 C 6.0 -0.7556189165 0.0000000000 0.8893840824 C 6.0 0.7556189165 0.0000000000 0.8893840824 H 1.0 0.0000000000 0.0000000000 -2.5225616018 H 1.0 -2.3139393369 0.0000000000 -0.5911050932 H 1.0 2.3139393369 0.0000000000 -0.5911050932 H 1.0 -1.3301790683 0.0000000000 1.8141412757 H 1.0 1.3301790683 0.0000000000 1.8141412757 Bq0 0.0 0.0000000000 0.0000000000 -0.0569023145 Bq1 0.0 0.0000000000 1.0000000000 -0.0569023145 $END
代表的な化合物の計算結果は以下の通りです(RHF(GIAO)/6-311G(d)//RMP2/6-31G(d))。芳香族は大きな負の値、反芳香族は大きな正の値を示しています。
| NICS(0) | NICS(1) | |
| Benzene | -10.35 | -12.24 |
| Pyrrole | -15.97 | -11.54 |
| Phosphole | -5.71 | -6.14/-5.77* |
| Cyclopentadiene | -3.57 | -5.28 |
| Borole | 17.34 | 9.64 |
| Cyclobutadiene | 28.98 | 20.12 |
*Phospholeは環の面に対して非対称のため、Lp側/P-H側それぞれの計算値
※1:ボルハルト・ショアー現代有機化学(日本語第2版), p.613
※2:Org. Lett. 2002, 2873.
※3:Tetrahedron. 2003, 1657.
※4:J. Am. Chem. Soc. 1996, 6317.
Leave a Reply
コメントを投稿するにはログインしてください。