非常に大きな分子を計算するとき、その計算時間を削減する方法としてQM/MM法が現在有力な方法の一つとなっています。大きな分子においては全ての原子が重要ではなく、多くの原子は立体的な影響のみで、電子状態が重要な寄与を果たすのは一部であるという考え方は、直感的に受け入れやすいものです。そういう場合、分子を「精密に計算する部分:高精度ab initio法やDFT」と「粗く計算する部分:低精度ab initio法や半経験法,分子力学」という風に区分して計算できれば、計算資源の節約につながります。
GaussianではONIOM法が実装されており、分子を最大3つのレイヤーに切り分け、各層に別々のモデル化学(非経験的分子軌道法および密度汎関数理論,半経験的分子軌道法,分子力学)を割り当てることができます。
一方、GAMESSではONIOM法の基となったIMOMM/SIMOMM法が実装されています。これは、分子を2つのレイヤーに切り分け、一方に量子力学計算を、もう一方に分子力学計算を割り当てます。分子力学のエンジンとしてはTINKERが使われています。ここでは、IMOMM法を使ったQM/MM計算の簡単な例を紹介します。
計算の下準備
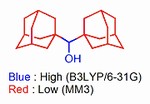
例として、Di(1-Adamantyl)methanol(DAM)のQM/MM計算を示します。GAMESSに於けるQM/MM法の入力には2つのパターン:QM部分を$DATAグループで明示する方法と$DATAグループを使わない方法がありますが、ここでは入力作成が簡単な後者で説明します。Fig.1に、DAMの平面構造式とQM/MM計算の切り分けを示します。2つのアダマンチル基は立体的な影響のみを考慮してMM3、コアのメタノール部分は高精度のB3LYP/6-31Gで計算します。
TINKERの入力ファイルを作成できるFacioでモデリングを行います。アダマンチル基の導入は2つのPDBファイルの結合により行います(Fig.2)。ここで、[Calculations>TINKER-MM3]でMM3計算を行います。すると、TINKERの実行ファイルと同じフォルダに「Facio.xyz」というTINKER形式のファイルが生成しますが(Fig.3)、この中身をコピーしてGAMESS用の入力ファイルを作成します。
入力ファイルの作成
QM/MM法の入力ファイルは、以下の形式になります。
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE COORD=TINKER
NZVAR=156 DFTTYP=B3LYP $END
$SYSTEM TIMLIM=600 MWORDS=1 $END
$BASIS GBASIS=N31 NGAUSS=6 $END
$STATPT NSTEP=500 $END
$ZMAT DLC=.T. AUTO=.T. $END
$TINXYZ
54 Diadamantylmethanol
1 C -0.406887 0.648500 -0.877586 1 2 3 8 31
2 O -0.167219 0.192571 -2.227781 6 1 4
3 H -0.877130 1.658578 -0.923399 5 1
4 H -0.527073 0.825385 -2.835354 21 2
5 C 2.979708 0.736553 1.991301 1 6 10 11 12
6 C 2.290529 2.064244 1.630505 1 5 7 13 14
(中略)
49 H -4.437918 0.374482 1.239451 5 41
50 H -2.743899 -0.097431 -2.097237 5 38
51 H -3.104916 1.084292 -0.812566 5 38
52 H -4.470824 -3.010351 -0.441549 5 46
53 H -3.542017 -2.509433 -1.882653 5 46
54 H -4.935088 -0.603425 -1.021576 5 48
$END
$LINK IMOMM=.T. IQMATM(1)=1,2,3,4 $END
$TINKEY
parameters C:\WinGAMESS\params\mm3.prm
$END
まずポイントとなるのは$CONTRLグループの「COORD=TINKER」で、これを指定することで$DATAグループはスキップされ、代わりに$TINXYZグループでTINKER形式の座標指定をします。$ZMATグループで内部座標自動発生を指定し、$TINXYZグループ内に、Facio.xyzからコピーしたTINKER形式の座標を入れます。
QM/MMの重要な指定が$LINKグループで、計算の方法としてIMOMM法(F.Maseras, K.Morokuma J.Comput.Chem. 16, 1170(1995).)を指定※1、IQMATM(1)で、QM法を用いる($CONTRL,$BASISによるモデル化学を適用する)原子を番号で指定します。切断部分は自動的に水素が補完されます。$TINKEYでパラメータファイルをフルパスで指定します。$TINKEYは上記のように一行一行改行する必要があります。
計算の実行と結果の抽出

計算はbatmaker.exeでbatファイルを作成して実行します。Pentium M 755(2.0GHz)で9.7分で終了しました。このoutファイルをFacioやWinmostarで読み込んでも、コア部分のメタノールしか表示されません。outファイルを開くと、「***** EQUILIBRIUM GEOMETRY LOCATED *****」の前に「Cartesian Coordinates of Atoms in Bulk Model」という部分があり、ここが全原子の座標になります(Fig.4)。ここをコピーし(Fig.4のように選択するとオーソドックスなxyz形式,両端まで選択するとTINKER xyz形式)、xyzファイルとして保存し、Facio(またはWinmostar)で開くことで最適化構造を拝むことができます(Fig.5)。

IMOMM法はキャップ原子(QM部分とMM部分の切断面にかぶせる原子で、通常は水素をよく使います)の構造パラメータが固定されていて、SIMOMMはキャップ原子の位置も含めて構造最適化できます。$DATAでQM部分を指定する時は、キャップ原子を後ろにまとめるように指定します。このとき、キャップ原子を水素以外に変えることもできます。
$DATAを使うときの注意点として、$LINKのIQMATM(1)=a,b,c…の指定を$DATAと整合性が取れるようにする必要があり、例えば$TINXYZで4番炭素,8番酸素をQMに指定する際、$DATAでは1番酸素,2番炭素となっていたならば、IQMATM(1)=8,4という風になります(数字を小さい順に4,8と並べて書くと、この対応が乱れて計算がうまくいきません)。
Leave a Reply
コメントを投稿するにはログインしてください。